Researchers have used fruit flies to decipher an unexplained connection between Alzheimer’s disease and a genetic variation, revealing that it causes neurons to die.
The findings from the WEHI-led team uncover a possible cause of neurodegeneration in the preclinical stages of Alzheimer’s disease and open the door for the future development of new treatments for cognitive diseases.
At a glance
A strong genetic link between Alzheimer’s disease and the mitochondrial TOMM40 gene has been noted for over a decade, but never explained.
Using a well-established fruit fly model of neurodegenerative disease, researchers have gained new insight into this link and how it leads to the death of neurons.
The research improves our understanding of the preclinical stages of cognitive diseases associated with ageing and pinpoints previously unexplored avenues for the development of new therapies.
Over 55 million people worldwide are believed to be living with Alzheimer’s disease or other forms of dementia. In Australia, there are up to 487,500 people living with dementia. There is no known cure for the disease, but early intervention can help prevent disease progression.
The new research enhances our knowledge of how cell death and quality control pathways are involved in neurodegeneration and reveals potential targets for early intervention in cognitive conditions.
Gap in dementia research
A strong genetic link exists between increased levels of the mitochondrial TOMM40 gene and Alzheimer’s disease, but the mechanisms underlying this phenomenon are largely unknown.
Dr Agalya Periasamy and Dr Jacqui Gulbis (L-R) used fruit flies to decode an unexplained link between the development of Alzheimer’s disease and a genetic variation that causes neurons to die.
This connection has been hard to untangle because this gene neighbours ‘The Alzheimer’s gene’ – ApoE4 – the strongest predictor of late onset Alzheimer’s disease.
But recent work has shown a genetic variation that causes over-production of TOMM40 can cause brain shrinkage, independent of the Alzheimer’s gene.
Lead researcher Dr Agalya Periasamy said the findings piqued the team’s curiosity, prompting them to investigate how too much TOMM40 causes the neurodegeneration underlying Alzheimer’s disease.
“If we can unpack this, we might be able to find a new way to intervene with the process to prevent neurons from dying,” Dr Periasamy said.
“Currently, we don’t have good treatments for Alzheimer’s and we urgently need new options.
“Our research offers a possible alternative avenue for development of much-needed therapeutic interventions for this devastating disease.”
Fruit fly model
The WEHI and Australian National University team used a common model of neurodegenerative disease, the fruit fly, to explore potential connections between elevated TOMM40 levels and Alzheimer’s.
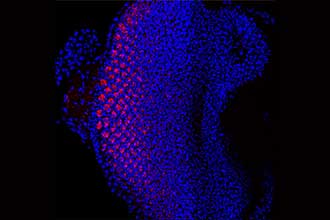
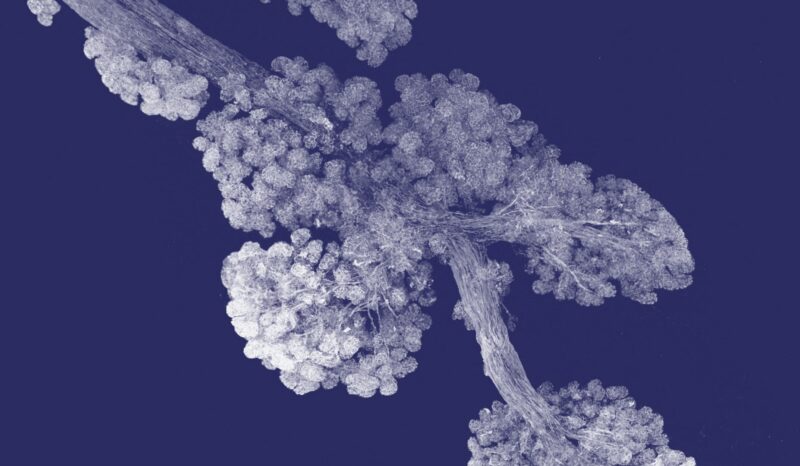
Eye of a fruit fly larva overexpressing the protein Tom40, which fluoresces red under the microscope. The new research helps explain the link between the gene that produces the Tom40 protein, TOMM40, and neurodegeneration.
The eyes of fruit flies contain cells called photoreceptors that are specialised neurons, making them ideal for research on neurodegeneration.
To investigate how an over-abundance of TOMM40 was linked to neurodegeneration, the team genetically engineered fruit flies to produce too much Tom40 protein, the protein produced by the TOMM40 gene, and observed the effect.
They found that enriching the protein caused marked cell death in the retina, with the amount of degeneration corresponding to the level of the protein.
After looking for the cause of the dying eye tissue, Dr Periasamy found evidence of a specific kind of cell death called apoptosis, typically involved in regular cell turnover and maintenance.
“We looked at the eyes of fly larvae under the microscope and found an increase in a protein that marks the activation of apoptosis, called ‘caspase-3’ in humans. This confirmed to us that apoptosis was the missing link we were looking for,” she said.
TOM protein complex and cell death
Tom40 is part of a larger protein complex called “TOM” that assembles on the mitochondria, where its main function is to import essential proteins.
The team investigated the impact of excessive Tom40 and found that TOM complex formation was sent into overdrive. This throws out the balance in the mitochondria and activates apoptosis.
“While the data show that the trigger for cell death is an overabundance of the TOM assembly, we found no evidence that protein import was involved in neurodegeneration,” said principal investigator Dr Jacqui Gulbis.
“Our findings identify a new entry point into cell quality control pathways that could be targeted to interrupt TOMM40-induced neurodegeneration.
“While this research is still in its early stages, it will be exciting to explore and tap into the relationship between TOMM40-linked apoptosis and Alzheimer’s disease to set the groundwork for the development of new therapies for cognitive conditions.”
The study, “An increase in mitochondrial TOM activates apoptosis to drive retinal neurodegeneration”, with collaborators from Australian National University, is published in Scientific Reports (DOI: 10.1038/s41598-022-23280-z).
WEHI authors: Agalya Periasamy, Peter Colman and Jacqueline Gulbis.